Energy exchange between adsorbates and surfaces

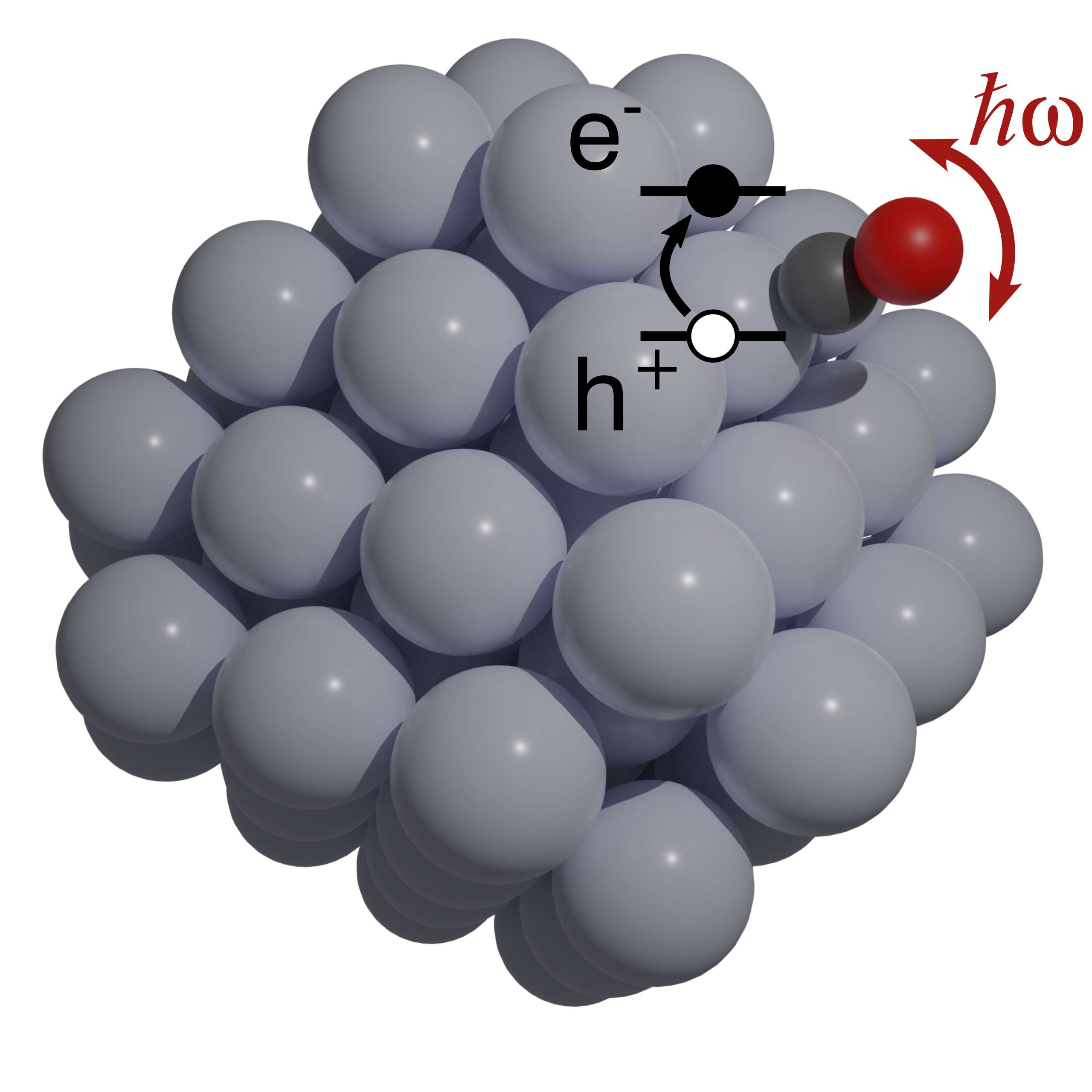
The interaction between surfaces and adsorbed molecules lies at the heart of many technologies: from catalysis and energy conversion to sensing and nanofabrication. The structure and stability of adsorbates determine whether a molecule binds weakly, diffuses, or activates toward a chemical reaction. At the same time, the exchange of energy between adsorbates and the surface (electrons, phonons) governs how efficiently reactions proceed, how long excited states persist, and how mechanical or thermal energy is dissipated at the atomic scale. By understanding and controlling these processes, we can reveal the microscopic rules that drive reactivity, selectivity, and functionality at surfaces, opening pathways to more efficient catalysts, durable materials, and novel quantum devices.