Projects
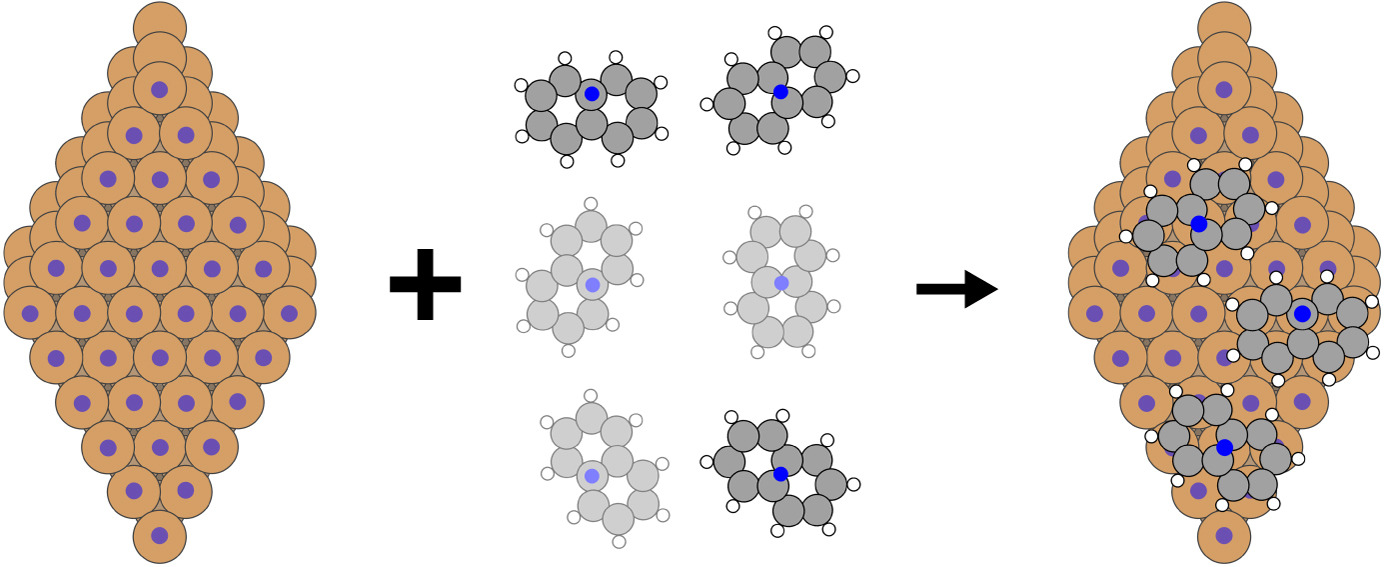
Structure and stability of defective 2D materials

Developing equivariant neural network potentials trained on first-principles data to perform high-throughput simulations of 2D materials. A particular focus lies on topologically designed defective graphene.
Ultra-fast energy exchange at surfaces

First-principles study of dynamic friction and electronic energy dissipation, supported by a Marie Skłodowska-Curie Fellowship.
Atomic-scale friction

Research atomic-scale frictional energy dissipation in collaboration with Jay Weymouth.
Surface structure prediction
Development of the SAMPLE structure prediction code. SAMPLE is the world's first quasi-deterministic structure prediction code for two-dimensional structures.
